Photoinitiated and Thermal Crosslinking of Epoxides Using Oxime-Blocked 2-Isocyanato-Ethyl-Methacrylate
Experimental
BMA and GMA were obtained from Aldrich Chemical Inc. and were purified by removing inhibitors prior to radical polymerization.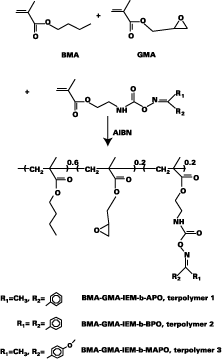
Preparation of different keto-oximes
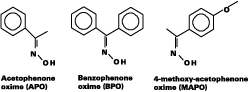
Acetophenone-oxime (APO) was prepared by refluxing acetophenone (20.0 g), pyridine (15.8 g) and hydroxylamine hydrochloride (13.9 g) in 200 mL ethanol. An analogous procedure was followed for the preparation of benzophenone oxime (BPO).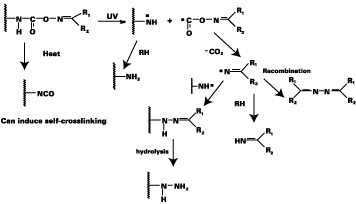
Preparation of the monomer IEM blocked with keto-oximes
IEM blocked with acetophenone oxime (IEMbAPO): IEM (3.0 g) and APO (2.7 g) were reacted in 25 mL toluene in the presence of 0.1 mL dibutyltin laurate. The reaction mixture was heated at 60 deg C for five hours. Subsequently, 0.5 mL isopropylglycidyl ether was added and the solution was stirred for another 30 minutes to scavenge possibly formed amine groups. IEM blocked with benzophenone oxime (IEMbBPO), and IEM blocked with 4-methoxy acetophenone oxime (IEMbMAPO) were prepared following analogous procedures.
Polymerization
The terpolymers of BMA, GMA and keto-oxime-blocked IEM were prepared in toluene using AIBN as the initiator following standard polymerization procedures. The chemical composition of the terpolymer, as determined by 1H NMR, was 60% BMA, 20% GMA and 20% keto-oxime-blocked IEM. Table I shows the molecular weights (GPC) and the lmax values (UV) for the polymers.Instrumentation
1H NMR spectra were recorded on a Bruker 400 MHz FT-NMR spectrophotometer using tetramethylsilane (TMS) as an internal standard. Ultraviolet spectra were obtained using a Hitachi U 2001 spectrophotometer. IR spectra were recorded on a Bio-Rad FTS 6000 FTIR spectrophotometer. The molecular weight of the polymers was analyzed using a Shimadzu LC-10AD VP liquid chromatograph equipped with a UV detector (Applied Biosystems 785A) and a mixed-D column (7.5 mm x 60 cm, Polymer Laboratory). Chloroform was used as the mobile phase. Polystyrene was used as the calibration standard.
Irradiation experiments
A solution of the terpolymer (5 wt%) in THF was prepared. Four drops of this solution were placed on a quartz plate and spread manually to obtain films of even thickness. After evaporation of the solvent, the polymer films were thoroughly dried in a vacuum oven at 40 deg C. The thickness of the films after drying was 5-10 mm. The quartz plate containing the polymer film was exposed to the UV light of an F-lamp in a UVACUBE (2000 W) irradiation instrument (Dr. Honle AG). During irradiation of the dry polymer film the distance between the sample and the UV source was maintained at 15 cm.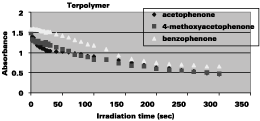
Thermal experiments
Thermal experiments were carried out in a convection oven at 50 to 150 deg C for different time periods. Thermal treatments of polymer films were performed either before or after the UV irradiation.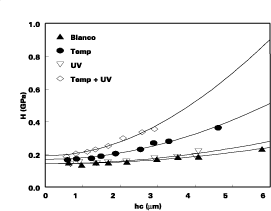
Nano-indentation experiment
The nano-indentation experiments were conducted at room temperature and ambient atmosphere using the procedure described by Malzbender et al.15,16. A Berkovich-type indenter was used to analyze the hardness and the reduced elastic modulus of the polymer films. The load ranged from 2 to 1000 mN. The diamond tip was inserted into the film with a speed of 10 nm/s until the maximum load was reached.Results and Discussion
IEM monomers having oxime-urethane moieties were prepared as shown in Scheme 1. These modified IEM materials were used to prepare the terpolymer using GMA and BMA as the other monomers (Scheme 2).BMA was used as the base monomer, which simultaneously reduces the glass transition temperature (Tg) of the polymer, thus maintaining a high mobility and allowing chemical reactions to take place. The epoxide groups supplied by the GMA monomer were the crosslinking sites for the in-situ-generated amine.
The photodecomposition supposedly takes place according to the mechanism shown in Scheme 3. Important factors in this mechanism are:
1. Irradiation of light leads to the homolytic cleavage of the N-O bond, which results in the formation of an iminyl and carbonate radical pair. This process is accompanied by the disappearance of the strong UV-band at ~250 nm that is present in the starting material.
2. An active amine and/or hydrazine is formed that is able to react with a second component in the mixture, such as an epoxy-(multi)functionalized material (Scheme 4), thus leading to crosslinking.
3. The irradiation leads to a release of carbon dioxide and a derivative of the parent ketone. Volatile materials will tend to evaporate, others will remain in the coating, but in both cases coating quality, health and environmental consequences will have to be faced.
The current study deals with the first issue, the photochemical decomposition of the blocked isocyanate, as witnessed by the disappearance of the corresponding UV-absorption band.
Figure 1 shows the UV spectra of terpolymer 1 (BMA-GMA-IEMbAPO) at different irradiation times. There is a gradual decrease in absorbance with UV irradiation time, indicating the disappearance of the chromophoric group. In the IR spectra, the decrease in intensity of the peaks at 1320 and 1170 cm-1 indicates the cleavage of the -N=C bond in the terpolymer. The same observation was made in case of terpolymer 2 (BMA-GMA-IEMbBPO) and terpolymer 3 (BMA-GMA-IEMbMAPO).
Figure 2 shows the change in absorbance with irradiation time for the different terpolymers. The absorbance decreases relatively fast in the first few seconds and then it levels off. The absorbance of the different terpolymers was determined at their lmax as given in Table 1.
It can be seen that the initial decomposition rate of the acetophenone derivatives is significantly higher than the one of the benzophenone compound, but that the overall process is rather slow, requiring minutes rather than seconds. In very recent studies17 we have seen that the decomposition rate of oxime-blocked isocyanates can be greatly enhanced by the use of specific photosensitizers, such as thioxanthones, leading to photodecomposition times of seconds.
Although the UV-spectra showed that decomposition had taken place, there was no sign of crosslinking for any of the terpolymers. When the polymer film was UV-irradiated after preheating to approximately 100 deg C, there was a remarkable change in the polymer film, leading to its (almost complete) insolubility in organic solvents. The same phenomenon was observed when the film was heated after the room temperature irradiation procedure.
The effect of crosslinking on the mechanical properties of the films, prepared from terpolymer 2 (BMA-GMA-IEMbBPO), was investigated by means of nanoindentation measurements. This technique15,16,18 was used to characterize the terpolymer before and after crosslinking. Figure 3 summarizes the effect of irradiation and thermal treatment on the penetration depth of the nano-indentor instrument's microtip into the polymer film. Since these measurements indicate no significant change in properties when the films are only irradiated at room temperature, they indicate there is no crosslinking occurring when the polymer film is only subjected to UV irradiation.
However, there is a clear increase in hardness when the polymer is only heated (i.e., without any irradiation) at 120 deg C. This is not unexpected since, at this temperature, a part of the blocked isocyanate moieties will start to thermally deblock upon generation of free isocyanate groups. The presence of such groups could indeed be proven by independent FTIR measurements, which reveal the presence of a peak at 2275 cm-1 attributed to the -NCO group. The increase of the mechanical properties of the film is undoubtedly due to crosslinking in the polymer film through self-crosslinking induced by the -NCO group as explained in Scheme 3.
Figure 3 also shows there is a more drastic increase in hardness in the polymer film when it is heated at 120 deg C, immediately followed by UV irradiation. This indicates that a combination of UV-irradiation and thermal treatment is the most effective way to crosslink the polymer in terms of speed, temperature and time. At this time it is not clearly understood why the sole treatment with UV irradiation does not lead to crosslinking. Still, the mobility of the polymer chains may play a role in explaining the phenomenon. DSC measurements indicate that the Tg of the terpolymer is 55 ¼C, which is still well above room temperature, but which is close to the temperature of the polymer during the irradiation procedure.
The results clearly indicate that the reaction of epoxides with the in-situ-generated amines is the rate-determining step in the crosslinking process of this one-component system. Tsunooka et al.14 and Chae et al.13 also reported that heating is necessary to induce the crosslinking in the UV-irradiated samples.
Conclusion and Outlook
We have shown with UV-spectroscopic measurements that a terpolymer having epoxide and oxime-blocked isocyanate side groups can be photochemically deblocked. However, the deblocked polymer does not undergo spontaneous crosslinking via the reaction of the in-situ-generated amines with the epoxy groups. Thermal treatment after or during UV irradiation is necessary to induce the crosslinking in the terpolymer. Nano-indentation experiments have been used to characterize the mechanical properties of the films before and after irradiation and/or thermal treatment.
The currently investigated systems can be regarded as model compounds for industrially more attractive systems that do not contain the IEM monomer. Presently, we are investigating systems based on combinations of commercially available isocyanate oligomers and epoxy resins (and other reactive components). The aim is to develop systems that are commercially attractive and can be crosslinked by irradiation only. In this context, the use of suitable polymerization catalysts is also under investigation.
Acknowledgements
The authors gratefully acknowledge Dr. D. Burget (Ecole Nationale Sup‚rieure de Chimie de Mulhouse) for helpful suggestions and Dr. Arjen Boersma for his help in carrying out the nano-indentation experiments. This work has been funded by the EU Fifth Framework Program, project no. GRD1-2000-25541 ("LOTEC").
For more information, e-mail Barteld de Ruiter, TNO Industrial Technology, at b.deruiter@ind.tno.nl.
This paper was presented at the 7th Nurnberg Congress, European Coatings Show, April 2003, Nurnberg, Germany.
References
1 Radiation Curing of Polymers II; Randall, D.R., Ed.; Royal Society of Chemistry: Cambridge 1991.2 Photopolymerization and Photoimaging, Science and Technology; Allen, N.S., Ed.: Elsevier Applied Science: Barking 1989.
3 i) Fouassier, J.P. Photoinitiation, Photopolymerization and Photocuring: Fundamentals and Applications; Hanser Publishers: Munich 1995.
ii) Shirai, M.; Tsunooka, M. Prog. Polym. Sci., 21, 1 1996.
4 Fr‚chet, J.M.M. Pure Appl. Chem., 64, 1239 1992.
5 Crivello, J.V. CHEMTECH, 624 1980.
6 Ito, H.; Ueda, M. J. Photopolym. Sci. Technol., 2, 1 1989.
7 Ito, K.; Shigeru, Y.; Kawata, Y.; Ito, K.; Tsunooka, M. Can. J. Chem., 73, 1924 1995.
8 Ito, K.; Nishimura, M.; Sashio, M.; Tsunooka, M. J. Polym. Sci. Part A: Polym. Chem., 32, 2177 1994.
9 Ito, K.; Nishimura, M.; Sashio, M.; Tsunooka, M. J. Polym. Sci. Part A: Polym. Chem., 32, 1796 1994.
10 Roescher, G.A.; de Ruiter, B. PCT World Patent Appl., 9858980 filed 19 June 1998.
11 Roescher, A.; de Ruiter, B. Proc. XXV FATIPEC Congr., Turin, 2000, Vol. 1, 175 2000.
12 Chae, K.H. Macromol. Rapid Commun., 19, 1 1998.
13 Chae, K.H.; Song, H.B. Polym. Bull., 40, 667 1998.
14 Tsunooka, M.; Tachi, H.; Asakino, K.; Suyama, K. Proc. RadTech Europe '99, Berlin, 405 1999.
15 Malzbender, J.; de With, G.; den Toonder, J.M.J. Thin Solid Films, 366, 139 2000.
16 Malzbender, J.; de With, G.; den Toonder, J.M.J. Thin Solid Fims, 372, 134 2000.
17 Singha, N.K.; Haarmann, M.; de Ruiter, B. Unpublished results.
18 Oliver, W.C.; Pharr, G.M. J. Mater. Res., 7, 1564 1992.
Looking for a reprint of this article?
From high-res PDFs to custom plaques, order your copy today!


